

こんにちは、せいです。未経験から遺伝子検査会社でNGSのデータ解析をしています。
遺伝子解析のためのツールをいくつかインストールしたけど、実際どういうことができるんだろう。
無料で利用できるデータを活用して、実際に遺伝子解析をしてみましょう。今回はToMMoというデータバンクの日本人データを活用してSNP解析をします。
NGSデータ解析未経験の私が、遺伝子検査会社でNGSのデータ解析ができるようになるまでの道のり(解析に必要なツールやそのツールのインストール方法、データの解析方法)を紹介していきます。
今回の投稿では、遺伝子解析のためのツール利用して、実際のSNP解析をご紹介します。手元にNGSデータがない方のために、誰でも無料で使用できる東北メディカルメガバンク(https://www.megabank.tohoku.ac.jp/)で公開されている日本人のデータを活用していきます。
日本人データの取得

東北メディカルメガバンク機構とは
東北メディカルメガバンク機構は、東日本大震災により甚大な被害を受けた被災地の医療の再生と地域医療の復興、そして大規模な医療情報化の流れに対応する新規医療の構築のために設立されました。
東北メディカル・メガバンク事業が実施している三世代・地域住民ゲノムコホート事業と、その成果によって構築されるコアバイオバンクは、次世代医療である個別化医療の発展に大きく貢献する礎になると考えられています。
特に、グローバルにゲノムデータを取得したバイオバンクはこれまでにありましたが、日本人のみにフォーカスしたデータバンクはこれまでほとんどありませんでした。東北メディカルメガバンクが保有する日本人データを解析することで、日本人が特になりやすい、特有の病気と遺伝子の関連が解明される可能性があります。
東北メディカルメガバンクでは、こうして蓄積された情報の一部が無料公開されています。
ToMMoのオープンアクセスなデータを取得する
では早速、東北メディカルメガバンクのオープンアクセスできる日本人のデータを取得しましょう。東北メディカルメガバンク機構(ToMMo)が公開している日本人のデータは、ToMMoのHP(https://www.megabank.tohoku.ac.jp/)から取得が可能です。
ToMMoでは数千人規模の全ゲノム解析を行い、リファレンスパネルを構築しています。このパネルをもとに一塩基バリアント(Single Nucleotide Variant:SNV)の位置情報、INDEL(挿入(insertion, IN)・欠失(deletion, DEL))の頻度情報、アレル頻度情報及びアレル数情報などをまとめたデータベースである、jMorp(Japanese Multi Omics Reference Panel)を公開しています。特に今回使用する日本人のマルチオミクスデータ(jMorp)はこちらから取得できます。
では、実際に今回使用する日本人の全ゲノムデータを取得しましょう。今回は、常染色体の日本人ヒトゲノムのSNVの情報を活用します。
まず、jMorpのHPへ移動し、”Genome Variation”(赤枠)をクリックします。

すると、検索画面のあるページに移動します。本来このページでは、特定の遺伝子に関する情報を検索するためのページですが、右上の”Downloads”(赤枠)をクリックすると全ゲノムデータを取得することができます。
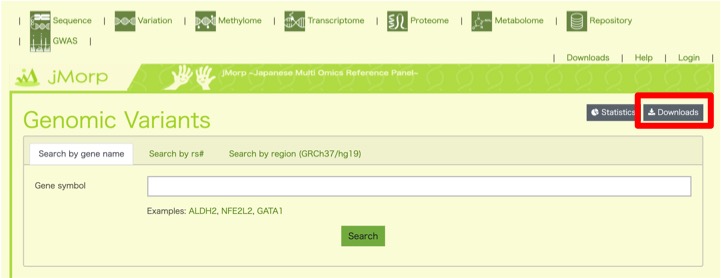
ヒトの全ゲノムデータを取得できるページに移動します。正確には、アレル頻度に関するデータに関しては無料でアクセスすることができます。遺伝子型の情報まで欲しい方は、ToMMoのユーザー登録が必要となります。
今回は、ヒトの全ゲノムデータから特定のSNPを抽出してくるため、無料公開されているアレル頻度に関するvcfファイルを取得します。常染色体のデータに関しては赤枠をクリックすると取得できます。
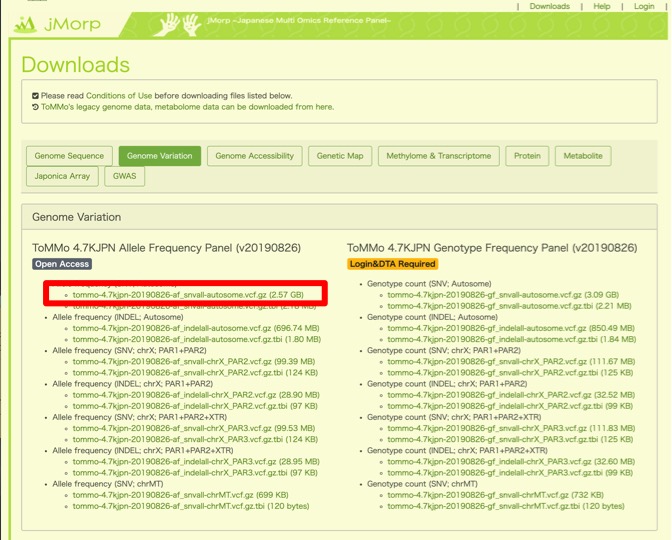
約3GBととても重いデータですので、PC本体ではなく外付けHDDにダウンロードすることをお勧めします。気長に待ちます。
また、
|
1 |
wget https://jmorp.megabank.tohoku.ac.jp/dj1/datasets/tommo-4.7kjpn-20190826-af_snvindelall/files/tommo-4.7kjpn-20190826-af_snvall-autosome.vcf.gz?download=true |
とコマンドを入力すれば、現在のディレクトリにファイルをダウンロードすることが可能です。
ダウンロードが完了したら、次の章でファイル内のデータを分割し、欲しいデータだけを取り出していきます。
vcfファイルから特定のデータを抽出する

特定の染色体のデータを抽出する
すべての常染色体のデータでは容量が大きい(約3GB)ので、実際にデータ解析をする場合は、染色体ごとに分割した方が扱いやすいです。いっぺんに全ゲノムを解析していことすると、使用しているパソコンのスペックによっては、解析に数時間かかったり最悪の場合解析途中でフリーズする可能性があります。
まずは、ヒトゲノムのすべての常染色体におけるvaliant callされたファイルから、特定の染色体だけのデータだけを抽出してきましょう。
ファイルが保存されているディレクトリに移動します。例えば、”DATA”という名称の外付けHDDにファイルがある場合は、
|
1 |
cd /Volumes/DATA/ |
で、目的のディレクトリに移動できます。実際にこのディレクトリにファイルがあるかどうかを確認する場合は、
|
1 |
ls |
と打ち込むと、現在いるディレクトリにあるファイルを確認することができます。
ここまでこれたら、早速vcfファイルから特定の染色体のデータだけを取り出しましょう。例えば、1番染色体のデータだけが欲しい場合は、
|
1 |
bcftools filter input.vcf.gz -r 1 > output.vcf.gz |
bcftoolsを使うことでヘッダーを残したまま特定の染色体のデータを抽出することができます。(入力ファイル名:input.vcf.gz、出力ファイル:output.vcf.gz のイメージです)
複数の染色体のデータを同時に抽出したい場合は、
|
1 |
bcftools filter input.vcf.gz -r 1,2,3 > output.vcf.gz |
で、1番染色体から3番染色体のデータをいっぺんに抽出することができます。
例えば今回取得したToMMoの常染色体のSNVのvcf.gzファイルから、1番染色体のデータのみを取り出したい場合は、
|
1 |
bcftools filter tommo-4.7kjpn-20190826-af_snvall-autosome.vcf.gz -r 1 > output.vcf.gz |
でできます。複数の染色体を抽出したい場合は、
|
1 |
bcftools filter tommo-4.7kjpn-20190826-af_snvall-autosome.vcf.gz -r 1,2,3 > output.vcf.gz |
とすると、1番染色体から3番染色体を同時に抽出することができます。
また、もしbcftoolsで上手くいかない場合は、grepでも特定の染色体のデータを抽出することができます。
|
1 |
grep -w '^#\|^1' input.vcf > output.vcf |
これで1番染色体だけのデータを抽出することができます。ただし、ヘッダーは無くなります。
いくつかの染色体を一緒に解析したい場合は、
|
1 |
grep -w '^#\|^[1-3]' input.vcf > output.vcf |
とコマンドすると、上記の場合は1番染色体から3番染色体の情報をいっぺんに抽出することができます。
もし、
|
1 |
Could not retrieve index file for 'ファイル名.vcf.gz' |
とエラーが表示されたらインデックスで解決します。
|
1 |
bcftools index ファイル名.vcf.gz |
このようにして特定の染色体だけを抽出したファイルを使用して、条件にあったSNPの情報を抽出していきましょう。
条件にあったデータを抽出す
次に、特定の染色体上にあるSNPの中でも、条件にあったSNPだけ抽出してきましょう。ここでは、vcffilterを使用します。
vcffilterのインストール方法は、【0からのNGSデータ解析】バイオインフォ初心者によるMacで遺伝子解析【準備編】をご覧ください。
まず、ヘッダーの意味を理解しましょう。
CHROM:SNPのある染色体の番号
POS:SNPのある位置
ID:SNPのID
REF:リファレンス配列における塩基
ALT:NGSなどで読みっとたリードの配列において、リファレンスと異なる塩基
QUAL:Quality scoreの値。検出ソフトや使用しているツールによって算出方法が異なる。
FILTER:フィルタリングが完了していれば”PASS”、していなければ”.”
INFO:追加情報。
・AC:allele count
・AF:allele frequency
・AN:total number of alleles
・DP(DEPTH):combined depth across samples
などなど。
例えば、ACが10以上、AFが30%以上のSNPが欲しい場合、
|
1 |
vcffilter -f "AC > 10 & AF > 0.3" input.vcf >output.vcf |
とすると、条件にあったSNPを抽出することができます。
また、拡張子をvcf→txtにすることで、Excelファイル上でも扱いやすくなります。もしコマンドが苦手という方は、Excel上でフィルタリングすることも可能です。
今回は、vcfファイルで特定の染色体上で条件にあったSNPを抽出する方法をご紹介しました。

コメント